

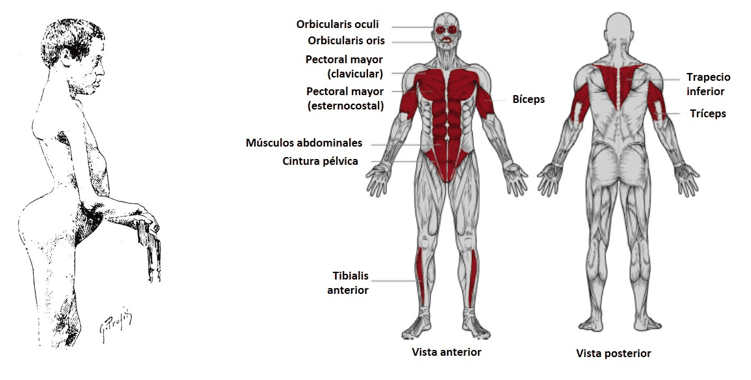
Ekainaren 20an muskulu-distrofia fazioeskapulohumeralaren Mundu Eguna ospatzen da. Gaixotasun hori eta Institutuan egiten den ikerketa ezagutzeko, Diagnostiko Molekularreko Plataformako arduradun den Pilar Camañorengana jo dugu.
2000. urteaz geroztik, 1 motako muskulu-distrofia fazioeskapulohumeralaren azterketa genetikoa egiten dugu (FSHD1), eta gaixotasun horretarako erreferentzia nazionaleko zentroa gara. Helduen artean hirugarren gaixotasun neuromuskular ohikoena da, Duchenneren distrofiaren eta distrofia miotonikoaren ondoren, 12:100.000ko prebalentziarekin. Distrofia hori modu autosomiko menderatzailean heredatzen da; horrek esan nahi du ondorengoek % 50eko aukera dutela heredatzeko, nahiz eta noizbehinkako kasuen (de novo) % 10-30en artean dagoen. Muskulu-distrofia bat da, lehen sintomak (debuta) erakusten dituena, normalean 10-20 urte bitartean, eta progresiboki eboluzionatzen duena, fazioeskapulohumeralako lurraldearen eragin asimetrikoarekin.
Akats genetikoa 4. kromosomaren beso luzean dago. Eskualde horretan, 3,3 kilobaseko tandemeko errepikapen genetikoz osatutako zati polimorfiko bat dago, D4Z4 izenekoa (1etik 150era bitarteko errepikapenak). Pertsona osasuntsu batek 10 D4Z4 errepikapen baino gehiago ditu, baina zati horrek 10 errepikapen baino gutxiago dituenean eta eremu horretan ingurune kromosomiko permisiboa dagoenean, pertsona batek FSHD1 du.
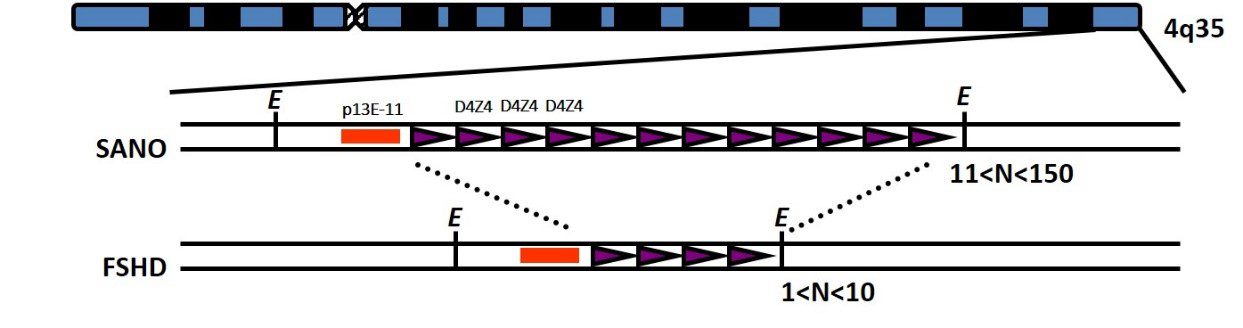
Gaixotasun horrek badu alde genetiko bat, baina badu beste zati epigenetiko bat ere, eta ingurune kromosomikoa –permisiboa esaten dioguna– eragiten duten zenbait berezitasun ditu. Horren ondorioz, azken errepikapenetan dagoen DUX4 genea egonkorra da eta transkribatu egiten da, eta proteina toxikoa da muskulu-zelularentzat. DUX4 genea aktibo dago enbrioi-garapenean, baina gero helduaroan isiltzen da, hau da, metilazioa izeneko mekanismo epigenetikoaren bidez haren funtzioa erreprimitzen da. Ezaugarri horiek gertatzen direnean (10 errepikapen baino gutxiago eta ingurune permisiboa), metilazioak ez du behar bezala funtzionatzen, eta, horren ondorioz, DUX4 genea aktibatu egiten da.
Bestalde, pazienteen % 5 negatiboak dira FSHD1erako, baina FSHD1en antzeko klinika dute. Paziente horiek normalean mutazioak izaten dituzte 18. kromosoman dagoen SMCHD1 genean. Gene horren funtzioa da lehen aipatu dugun 4 kromosomaren D4Z4 eremua aktibatzea saihestea. Gene horretan mutazioak daudenean, ezin du bere funtzioa bete, DUX4 genea aktibatuz eta FSHD2 sortuz. SMCHD1 genearen eta FSHD genearen arteko lotura hori 2012an aurkitu zen.
Laborategian FSHD1aren azterketa genetikoa egiten dugu, oso eskuzko metodo neketsu baten bidez (DNAren erauzketa, DNAren digestioa, elektroforesi horizontala, Southern blot, zundaren hibridazioa eta markatzea, mintzen garbiketa eta errebelatzea), eta 2014tik FSHD2aren azterketa genetikoa ere egiten dugu.
FSHDrako erreferentzia nazionaleko zentroa izateak aukera eman digu FSHD1erako 6.000 paziente baino gehiago aztertzeko, bai ospitaleetakoak, bai zentro pribatu nazionaletakoak, baina baita nazioartekoak ere (Belgika, Finlandia, Frantzia, Erresuma Batua, Irlanda, Malta, Portugal, Iran, Israel, Turkia, Zeelanda Berria, Peru, Los Angeles –AEB–). Ia % 50 izan dira positiboak; eta 250 paziente baino gehiago FSHD2rentzat, kasuen % 38 positiboak izanik.
Laborategian FSHDa aztertzen hasi ginenetik, hainbat ikerketa-proiektutan parte hartu dugu, eta lankidetza-lanak egin ditugu FSHDn puntakoak diren nazioarteko beste talde batzuekin. Lan horien emaitza 11 argitalpen garrantzitsu izan dira, azterketa genetikoari eta gaixotasun horrek bere mekanismo patologikoetan dituen berezitasunei dagokienez.
Gaur egun, FSHDak ez du sendabiderik, baina azken urteotan gero eta saiakuntza kliniko gehiago egin dira molekula batzuk probatzeko, eta molekula horien helburu terapeutikoa DUX4 genea da; izan ere, lehen esan dugun bezala, toxikoa da muskuluarentzat. Saiakuntza kliniko horiei begira, oso garrantzitsua da pazienteek beren azterketa genetikoa izatea eta ondo karakterizatuta egotea. Eta, horretarako, oso garrantzitsua da FSHDko pazienteen erregistro nazionala izatea. 2022ko urtarriletik ari gara erregistro horretan lanean (FSDH-Spain Elkartearen Batzorde Zientifikoaren barruan gaude), eta erregistro hori, azkenean, 2024ko azarotik dago martxan.
Beste alde batetik, FSHD-Spain aipatu nahi dugu, FSHD duten pazienteen elkartea, duela gutxi sortu dena (2017an hasi zen ibiltzen), eta haren kongresu nazionaletan parte hartu dugu, 2018an Burgosen egin zen lehenengotik 2025eko apirilaren 4-5ean Madrilen egin den azkenera arte.

Patologia hori duten pazienteen diagnostikoan lagundu ahal izatea oso garrantzitsua da, eta balio eta sari pertsonal gehigarria ematen dio gure lanari.

Argitalpenak
- Ehrlich M, Jackson K, Tsumagari K, Camano P, Lemmers RJ. Hybridization analysis of D4Z4 repeat arrays linked to FSHD. Chromosoma 2007 Apr;116(2):107-16.
- Sistiaga A, Camaño P, Otaegui D, Ibáñez B, Ruiz-Martinez J, Martí-Massó JF, López de Munain A. Cognitive function in facioscapulohumeral dystrophy correlates with the molecular defect. Genes Brain Behav. 2009 Feb;8(1):53-9
- Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010 Sep 24;329(5999):1650-3
- de Greef JC, Lemmers RJ, Camaño P, Day JW, Sacconi S, Dunand M, van Engelen BG, Kiuru-Enari S, Padberg GW, Rosa AL, Desnuelle C, Spuler S, Tarnopolsky M, Venance SL, Frants RR, van der Maarel SM, Tawil R. Clinical features of facioscapulohumeral dystrophy 2. Neurology 2010 Oct 26;75(17):1548-54
- Sacconi S, Camaño P, de Greef JC, Lemmers RJ, Salviati L, Boileau P, Lopez de Munain Arregui A, van der Maarel SM, Desnuelle C. Patients with a phenotype consistent with facioscapulohumeral muscular dystrophy display genetic and epigenetic heterogeneity. J Med Genet. 2012 Jan;49(1):41-6.
- Lemmers RJ, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, Camano P, Ramos Arroyo MA, Jerico I, Rogers MT, Miller DG, Upadhyaya M, Verschuuren JJGM, Lopez de Munain A, BGM van Engelen BGM, Padberg GW, Sacconi S, Tawil R, Tapscott SJ, Bakker B, van der Maarel SM. Inter-individual Differences in CpG Methylation at D4Z4 Correlate with Clinical Variability in FSHD1 and FSHD2. Hum Mol Genet. 2015 Feb 1;24(3):659-69.
- van den Boogaard M, Lemmers RJ, Camano P, van der Vliet PJ, Voermans N, van Engelen BG, López de Munain A, Tapscott SJ, van der Stoep N, Tawil R, van der Maarel Double SMCHD1 variants in FSHD2: the synergistic effect of two SMCHD1 mutations on D4Z4 hypomethylation and disease penetrance in FSHD2. Eur J Hum Genet. 2016 Jan;24(1):78-85
- Fernández-Torrón R, de Munain AL, Camaño P, García-Bragado F. Rapidly Reversible Winging Scapula. Arthritis Rheumatol. 2015 Sep;67(9):2502. doi: 10.1002/art.39202.
- Giacomucci G, Monforte M, Diaz-Manera J, Mul K, Fernandez Torrón R, Maggi L, CM Bettolo, Dahlqvist JR, Haberlova J, Camaño P, Gros M, Tartaglione T, Cristiano L, Gerevini S, Calandra P, Deidda G, Giardina E, Sacconi S, Straub V, Vissing J, Van Engelen B, Ricci E, Tasca G. Deep phenotyping investigation of FSHD2 by MRI: refining the FSHD-related disease spectrum. Eur J Neurol. 2020 Dec;27(12):2604-2615.
- Montagnese F, de Valle K, Lemmers RJLF, Mul K, Dumonceaux J, Voermans N; 268th ENMC workshop participants. 268th ENMC workshop – Genetic diagnosis, clinical classification, outcome measures, and biomarkers in Facioscapulohumeral Muscular Dystrophy (FSHD): Relevance for clinical trials. Neuromuscul Disord. 2023 May;33(5):447-462. doi: 10.1016/j.nmd.2023.04.005.
- Giardina E, Camaño P, Burton-Jones S, Ravenscroft G, Henning F, Magdinier F, van der Stoep N, van der Vliet PJ, Bernard R, Tomaselli PJ, Davis MR, Nishino I, Oflazer P, Race V, Vishnu VY, Williams V, Sobreira CFR, van der Maarel SM, Moore SA, Voermans NC, Lemmers RJLF. Best practice guidelines on genetic diagnostics of facioscapulohumeral muscular dystrophy: Update of the 2012 guidelines. Clin Genet. 2024 Apr 29. doi: 10.1111/cge.14533.





